
—— Wspólne badanie przeprowadzone przez Zhejiang CDC, Macro & Micro-Test i China CDC opublikowane w czasopiśmie Frontiers in Cellular and Infection Microbiology
Przegląd badań
W maju 2026 roku czasopismo Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4,6) opublikowało artykuł pod kierownictwem Centrum Kontroli i Prewencji Chorób Prowincji Zhejiang (Zhejiang CDC), którego współautorami byli zespół bioinformatyczny z Beijing Macro & Micro-Test Bio-Tech Co., Ltd. oraz Narodowy Instytut Kontroli i Prewencji Chorób Zakaźnych (China CDC). Badanie nosi tytuł:
„Identyfikacja i analiza filogenetyczna siedmiu szczepów Brucella abortus w Zhejiang w Chinach”.

Niniejsze badanie stanowi pierwszą systematyczną, opartą na całym genomie analizę identyfikowalności filogenetycznej Brucella abortus (B. abortus) w prowincji Zhejiang w Chinach. Zespół przeanalizował siedem izolatów zebranych w latach 2015–2025 (cztery szczepy pochodzenia ludzkiego i trzy szczepy pochodzenia bydlęcego z Jinhua, Quzhou i Ningbo). Odkrycia dostarczają genomicznych dowodów na pochodzenie i drogi transmisji tego „dominującego gatunku północnego” w nietypowym południowym regionie epidemicznym we wschodnich Chinach.
Tło i znaczenie
Bruceloza to choroba odzwierzęca wywoływana przez bakterie z rodzaju Brucella. Brucella abortus atakuje głównie bydło, ale może również powodować choroby u ludzi. W Chinach bruceloza charakteryzuje się znacznym zróżnicowaniem geograficznym: najwyższa zapadalność występuje w prowincjach północnych (np. Mongolia Wewnętrzna, Shanxi, Heilongjiang). Z kolei prowincje południowe, w tym Zhejiang, historycznie były zdominowane przez Brucella melitensis, z bardzo nielicznymi odnotowanymi przypadkami B. abortus. Ta regionalna dysproporcja sprawia, że charakterystyka genetyczna i śledzenie źródeł B. abortus w Zhejiang stanowią kluczowy priorytet zdrowia publicznego.
Metody i kluczowe ustalenia
Zespół badawczy przyjął wielotorową strategię łączącą biologię molekularną i bioinformatykę:
1.Identyfikacja patogenów i podstawowe typowanie
Badanie PCR genu BCSP-31 i AMOS-PCR potwierdziło, że wszystkie siedem izolatów to B. abortus.
Wielogenowe typowanie sekwencji (MLST) na podstawie dziewięciu genów gospodarczych wykazało, że wszystkie izolaty należały do typu sekwencji ST2, co wskazuje na wysoką jednorodność genetyczną wśród szczepów B. abortus krążących w Zhejiang.
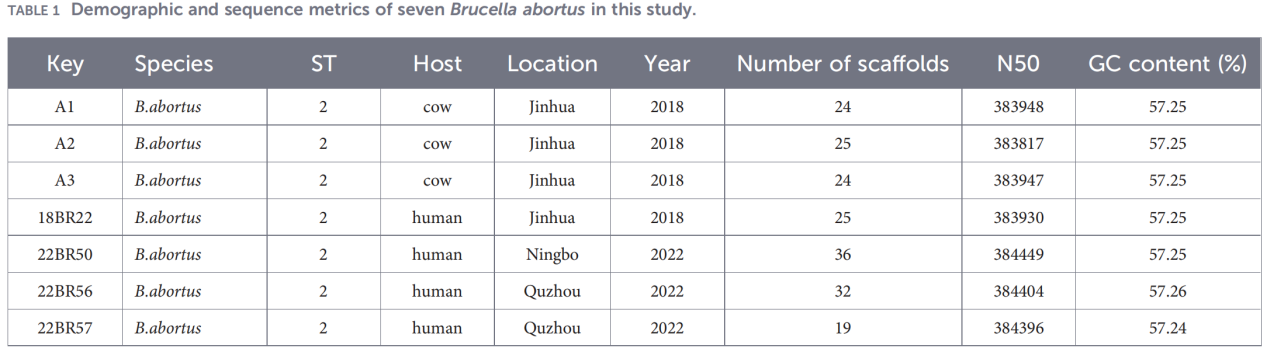
2.Charakterystyka całego genomu
Sekwencjonowanie całego genomu przeprowadzono na platformie Illumina NovaSeq. Analiza średniej identyczności nukleotydów (ANI) wykazała, że izolaty Zhejiang wykazują do 99,99% podobieństwa do szczepu referencyjnego B. abortus 544.
Analiza całego genomu wykazała, że populacja jest wysoce konserwatywna: zidentyfikowano 3084 geny rdzeniowe i tylko 10 genów powłoki, nie wykryto natomiast żadnych genów rdzenia miękkiego ani genów chmur.
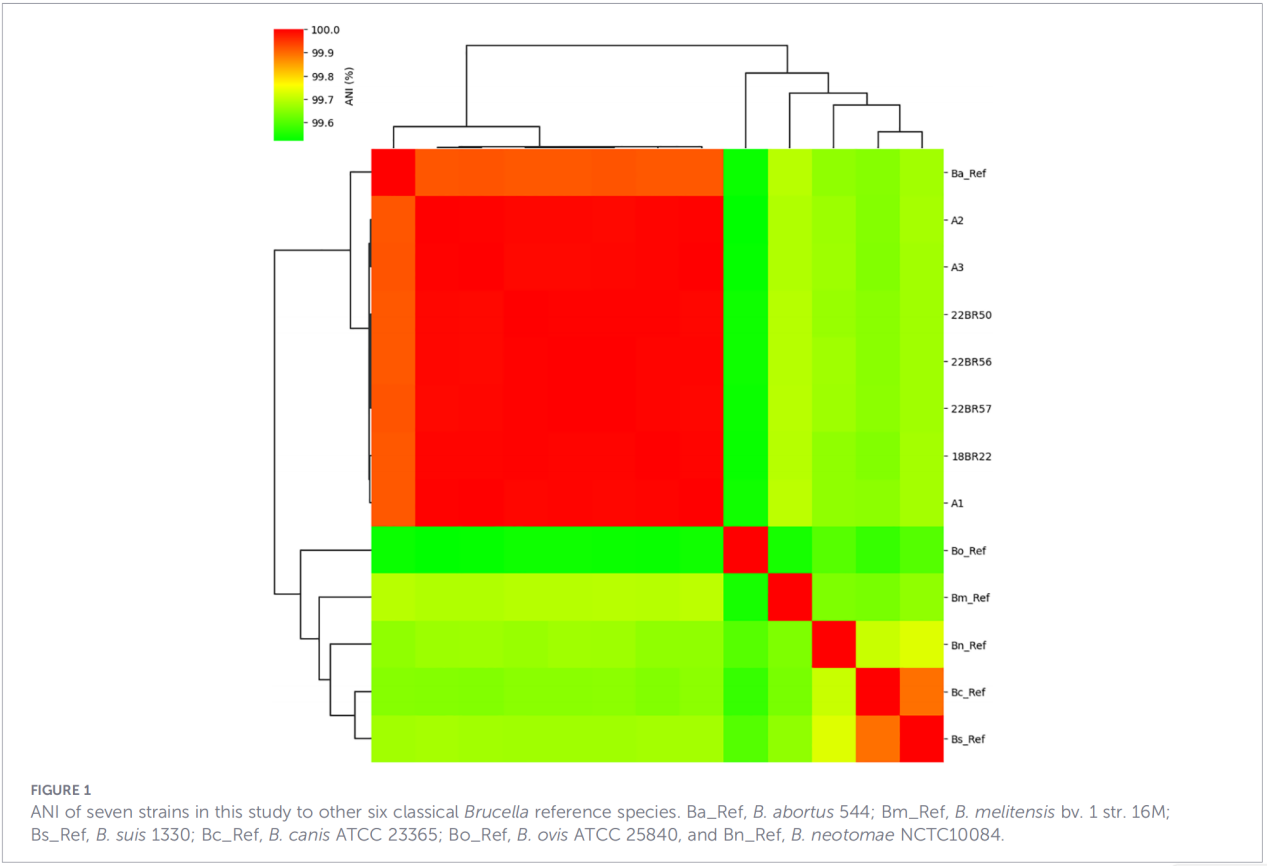
3.Profile genów wirulencji i oporności na środki przeciwdrobnoustrojowe
Przewidziano łącznie 68 czynników związanych z wirulencją, obejmujących klasyczne szlaki, takie jak biosynteza LPS, system sekrecji T4SS oraz dwuskładnikowy system regulacji BvrR-BvrS. Co istotne, wszystkie izolaty nie posiadały genów adhezyny bmaA i btaF. Analiza genu oporności wykryła jedynie gen mprF w bazie danych CARD, nie zidentyfikowano żadnych innych determinantów oporności.
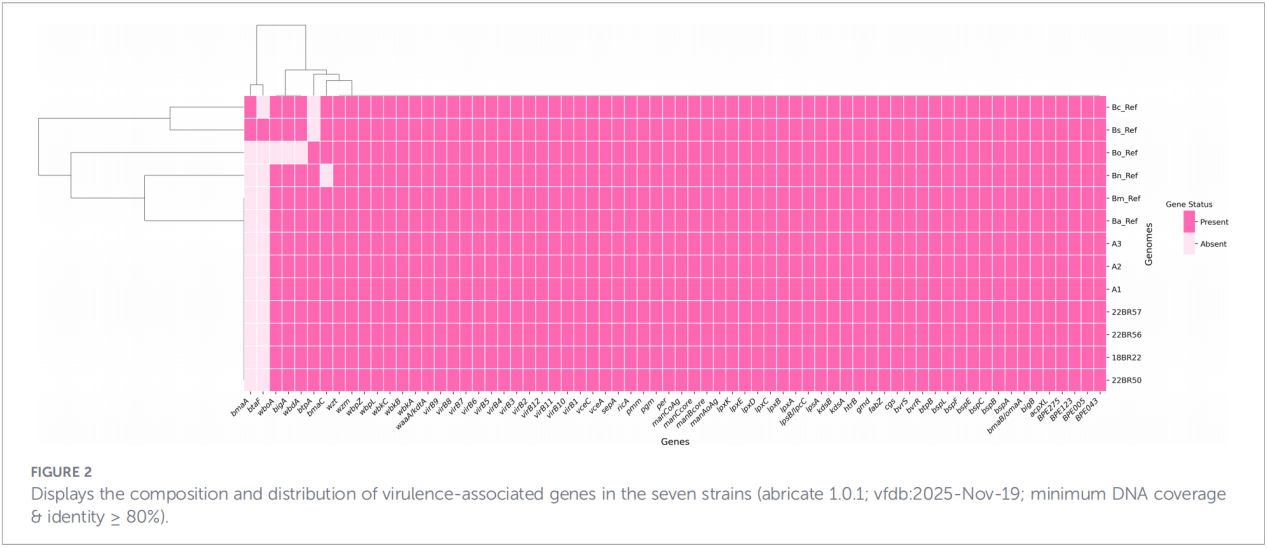 4. Rekonstrukcja filogenetyczna i śledzenie transmisji
4. Rekonstrukcja filogenetyczna i śledzenie transmisji
Analiza polimorfizmu pojedynczego nukleotydu (cgSNP) genomu rdzeniowego umiejscowiła izolaty Zhejiang w określonym miejscu na globalnym drzewie filogenetycznym. Wyniki pokazały, że szczepy Zhejiang tworzą grupę monofiletyczną wraz ze szczepami z Rosji, Mongolii i kilku północnych prowincji Chin (Ningxia, Heilongjiang, Mongolia Wewnętrzna, Hebei, Gansu, Pekin). Grupa ta dzieli się następnie na trzy odrębne podklady (klady 1–3), co sugeruje liczne, niezależne procesy introdukcji.
Wnioski i implikacje
Badanie to dostarcza pierwszego precyzyjnego zbioru danych genomicznych dotyczących B. abortus w prowincji Zhejiang i pozwala wyciągnąć kilka kluczowych wniosków:
- Klejar tło genetyczne– Szczepy B. abortus krążące w Zhejiang należą do ST2, są wysoce konserwatywne genomowo i stanowią typowy szczep brucelozy bydła.
2. Evistopień transmisji międzyregionalnej– Analiza filogenetyczna nie potwierdza istnienia niezależnej linii endemicznej w Zhejiang. Dane silnie sugerują natomiast, że szczepy te pochodzą z północnych Chin i mogą mieć wspólne pochodzenie ewolucyjne ze szczepami z Rosji i Mongolii. Obecność trzech podkladów sugeruje wiele odrębnych zdarzeń introdukcyjnych.
3. Konsekwencje dla zdrowia publicznego– Odkrycia podkreślają wartość nadzoru genomicznego nad brucelozą, nawet w regionach tradycyjnie nieendemicznych, takich jak Zhejiang. Chociaż obecna liczba przypadków jest niska, narzędzia o wysokiej rozdzielczości, takie jak cgSNP, mogą skutecznie namierzyć źródło importowanych ognisk choroby i dostarczyć dowodów naukowych umożliwiających przerwanie łańcuchów transmisji związanych z transportem zwierząt gospodarskich między prowincjami.
Praca ta nie tylko wypełnia lukę badawczą w prowincji Zhejiang, ale także dostarcza nowych danych bazowych do monitorowania patogenów i oceny ryzyka brucelozy w regionie delty rzeki Jangcy.
Informacje o artykule:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … i Wu, B. (2026). Identyfikacja i analiza filogenetyczna siedmiu szczepów Brucella abortus w Zhejiang w Chinach. Frontiers in Cellular and Infection Microbiology, 16, 1758-965.
Czas publikacji: 10 czerwca 2026 r.